
 Discussion
Discussion
Causes of Fabry disease
Last updated July 11, 2024, by Lindsey Shapiro, PhD
 Fact-checked by Ana de Barros, PhD
Fact-checked by Ana de Barros, PhD
Fabry disease is a rare genetic disorder caused by mutations in the GLA gene, which lead to an accumulation of fatty molecules, mainly globotriaosylceramide (Gb3), within cells.
This accumulation results in progressive organ damage, particularly to the heart, kidneys, and nervous system, consequently leading to the diverse range of Fabry disease symptoms.
As an inherited condition, Fabry disease runs in families and is passed down from parents to their children. Due to the location of the GLA gene in a sex chromosome, the disease’s symptoms may differ between male and female patients. Still, both men and women can be affected by Fabry disease and transmit it to their offspring.
What causes Fabry disease?
Fabry disease is caused by mutations in a gene called GLA, which provides cells with the instructions for making the alpha-galactosidase A (alpha-Gal A) enzyme. This enzyme works to break down fatty molecules like Gb3 inside lysosomes, the cell compartments that are responsible for recycling cellular waste.
In people with Fabry, there’s not enough functional alpha-Gal A being produced to break down Gb3 and related molecules. As a result of Fabry disease enzyme deficiency, those molecules accumulate to toxic levels in cells throughout the body and damage organs.
The etiology of Fabry disease — its underlying cause — places Fabry into a class of conditions known as lysosomal storage disorders. These are broadly characterized by the abnormal accumulation of toxic substances inside cells due to enzyme deficiencies that impair lysosomal function.
Nearly 1,000 Fabry-causing mutations have been found in the GLA gene, and different mutations can have differing effects on alpha-Gal A production. Some lead to completely absent or very low alpha-Gal A activity (less than 1% of normal) that’s the cause of classic Fabry disease, a more severe disease form that usually develops in childhood or adolescence. In other cases, the alpha-Gal A that’s produced has some residual activity (more than 1%), leading to the more common late-onset Fabry disease that emerges in adulthood.
Currently, there are no other known Fabry disease causes, and no additional Fabry disease risk factors for developing the condition have been identified.
However, the severity of Fabry symptoms and disease progression can vary among individuals based on factors such as the specific mutation, genetic and environmental modifiers, and individual patient characteristics.
Inheritance
The genetic basis of Fabry disease was identified in the late 20th century. While scientists discovered that alpha-Gal A deficiency is what causes Fabry disease in the 1960s, it wasn’t until the 1990s that they pinpointed the GLA gene as the specific gene responsible for this deficiency.
The GLA gene resides on the X chromosome, which is one of the two sex chromosomes (the other being Y). Females have two X chromosomes — one inherited from each biological parent — while males have one X chromosome inherited from their biological mother and one Y chromosome from their biological father.
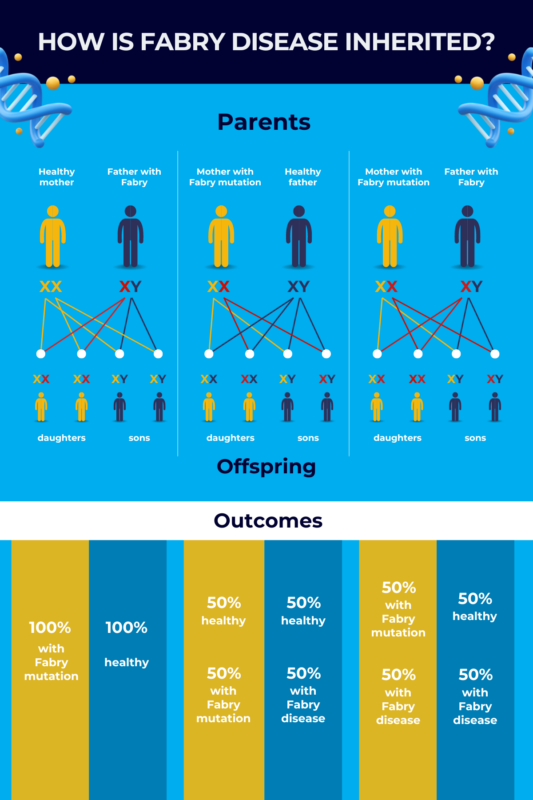
Fabry is passed down from a parent to a child in a X-linked dominant manner. This Fabry disease inheritance pattern means that a person has to inherit only one mutated copy of the GLA gene on a single X chromosome for the disease to manifest.
If a man with Fabry disease has children with a woman who does not have the condition:
- all female children will carry a mutation in one of their X chromosomes, as men pass their sole X chromosome on to all of their daughters.
- no male children will inherit the disease, as men only give a Y chromosome to sons.
Women pass on an X chromosome to all of their children, regardless of sex. As such, when a female with one mutated GLA gene copy has children with a man without Fabry disease, there is a 50% chance of any child — male or female — carrying a mutated gene.
If a man and woman each with one mutated GLA gene copy have children, there’s a 50% chance that their sons will have the disease. All of their daughters will have at least one mutated GLA gene, and there is a 50% chance that a daughter will inherit two mutated copies of the gene. This is extremely rare, but should it occur, all of her future children would inherit a Fabry mutation.
Given the established Fabry disease genetic causes, a person who has a Fabry disease family history may be at risk for developing it. They should be tested to ensure a prompt Fabry diagnosis and initiation of disease treatments.
In rare cases, spontaneous, or de novo, Fabry-causing mutations have been observed in patients without a family history of the disease. If a person has Fabry due to a de novo mutation, they still can pass it on to their children.
Differences between males and females
As an X-linked genetic disorder, Fabry disease can affect male and female patients differently.
Because males have only one X chromosome, a Fabry-causing mutation on their GLA gene typically leads to a more standard and severe disease course. For females who inherit a Fabry mutation on one copy of their GLA gene, the healthy copy on their other X chromosome may partly compensate for the mutated one, so the disease in females can be much more variable.
That variability likely is due to a process called X-chromosome inactivation. In early embryonic development, each cell of the female body randomly “turns off” one of its X chromosomes to avoid duplication of all the genes on that chromosome. Essentially, this means that some cells in the female body have active genes from the X chromosome inherited from the mother, while others have active genes from the chromosome inherited from the father.
In the context of Fabry disease, this means that some cells in a woman’s body will retain activity in the mutated copy of GLA and will produce the faulty alpha-Gal A protein, while others will have the healthy version and make a normal enzyme.
As the inactivation process is random, the type, severity, and timing of Fabry disease symptoms will vary depending on how many cells have the faulty GLA version in each tissue. Some female patients may go their entire lives without ever experiencing symptoms, and others may have a disease similar in severity to their male counterparts.
Prevalence
Fabry disease is found in both men and women across all racial and ethnic groups. The rare condition is currently estimated to affect around 1 in 1,000 to 9,000 people worldwide, but the exact prevalence of Fabry disease is not well established and estimates vary widely.
Late-onset Fabry is estimated to affect about 1 in 1,000 to 3,000 males and 1 in 6,000 to 40,000 females globally. In turn, classic Fabry disease is thought to affect around 1 in 22,000 to 40,000 males, whereas its prevalence in women is not known.
Fabry disease prevalence also may vary by geographic region, which can be influenced by differences in access to healthcare, diagnostic testing, and disease awareness.
It is now recognized that Fabry disease is underdiagnosed, and its prevalence could be higher than previously thought. This may be especially true in females, who until recently were largely considered not to be affected based on what was known about Fabry disease genetics. Advances in newborn screening programs and greater disease awareness may enable more patients to reach accurate and early diagnoses.
Fabry Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion