
Types of Fabry disease
Last updated Aug. 12, 2024, by Lindsey Shapiro, PHD
 Fact-checked by Ana de Barros, PhD
Fact-checked by Ana de Barros, PhD
Fabry disease, a rare genetic disorder, can be classified into two main types — classic and late-onset — that differ in terms of age at symptom onset, range of symptoms, and the rate of disease progression.
Both types of Fabry disease have the same underlying cause, that is, mutations in the GLA gene that result in a faulty or deficient alpha-galactosidase A (alpha-Gal A) enzyme. This leads to a progressive buildup of globotriaosylceramide (Gb3), a fatty substance that’s normally broken down by alpha-Gal A, resulting in organ damage and a range of symptoms throughout the body.
Around 1,000 mutations in the GLA gene have been reported to cause Fabry disease. As each mutation can affect alpha-Gal A production differently, patients will have varying levels of working enzyme in their bodies. Differences in how Fabry disease types manifest are explained by the amount of alpha-Gal A enzyme function patients retain.
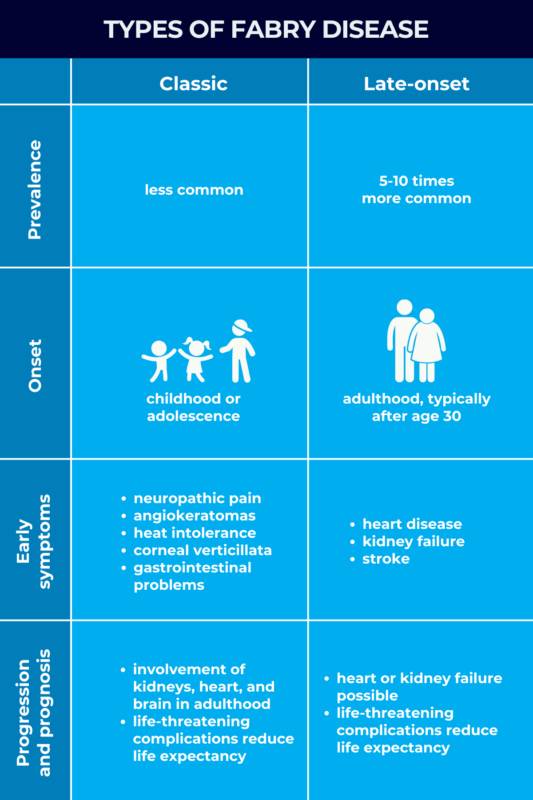
In classic Fabry disease, there is less functional enzyme, resulting in symptoms manifesting in childhood and progressing faster. In late-onset Fabry disease, the more common type, higher residual alpha-Gal A function results in symptoms starting in adulthood and progressing more slowly.
Regardless of disease type, Fabry disease is a progressive condition that can lead to life-threatening organ damage. An early Fabry disease diagnosis and start to treatment is important for ensuring the best possible outcomes for both classic and late-onset Fabry patients.
Classic Fabry disease
In classic Fabry disease, also known as type 1 Fabry disease, GLA gene mutations result in little to no alpha-Gal A activity (less than 1% of normal), leaving the body largely unable to clear Gb3. This leads to a significant accumulation of the fatty molecule from a young age, resulting in a generally more severe disease course.
Classic Fabry is the less common of the two disease types, affecting an estimated 1 in 22,000 to 40,000 males. Its prevalence in female patients is not established.
The earliest symptoms of classic Fabry disease usually emerge in childhood or adolescence, with some patients having noticeable symptoms as early as age 2. Early Fabry disease symptoms commonly seen in the classic form include:
- neuropathic pain characterized by burning sensations, especially in the hands and feet
- angiokeratomas, or clusters of dark red spots on the skin
- corneal verticillata, a starburst-like pattern of cloudiness on the surface of the eye
- reduced ability to sweat that causes heat intolerance
- gastrointestinal problems
- delayed puberty or delayed growth.
As children with classic Fabry age, the progressive accumulation of Gb3 eventually leads to more serious organ involvement in adulthood, including:
- deterioration of kidney function and eventual kidney failure
- heart disease, including abnormal heart rhythms, problems with the heart’s pumping muscle, and heart failure or heart attack
- cerebrovascular disease, or problems with blood flow in the brain, such as stroke and transient ischemic attacks (ministrokes).
Because of the increased risk of these potentially life-threatening health problems, Fabry disease life expectancy tends to be shorter than the average observed in the general population. If left untreated, men typically live to their late 50s, whereas women may live into their 70’s.
For that reason, Fabry disease treatment, including enzyme replacement therapy (ERT) or chaperone therapy, should be started as early as possible in children with classic Fabry disease to help slow disease progression and delay or prevent life-threatening organ damage.
Late-onset Fabry disease
Late-onset Fabry disease is a milder form of Fabry that, unlike the classic type, usually doesn’t manifest until after age 30.
People with late-onset, or atypical, Fabry disease have greater residual alpha-Gal A enzyme activity than people with classic Fabry (more than 1% of normal). While some Gb3 can be broken down naturally in the body, this fatty molecule will still accumulate, albeit at a slower rate. This means that disease progression is slower.
Late-onset Fabry is the most common form of the disease — at least 5-10 times more common than classic Fabry — affecting about 1 in every 1,000 to 3,000 men and 1 in 6,000 to 40,000 women worldwide.
Aside from starting later in life and progressing at a slower rate, late-onset Fabry disease symptoms also are different from those seen in the classic form of the disease.
Early symptoms of classic Fabry, such as neuropathic pain and angiokeratomas, are not as common in the late-onset form. Instead, the first indication of the condition may be problems with a major organ system, including:
- heart disease
- kidney failure
- cerebrovascular disease, such as stroke.
Late-onset Fabry disease is still a progressive condition, and major organ involvement can give rise to life-threatening complications like kidney or heart failure just as in classic Fabry. Late-onset Fabry disease symptoms may also be more easily missed by doctors because patients lack some of the hallmark early signs seen in classic Fabry disease. This can delay treatment and lead to avoidable complications.
As with classic Fabry disease, the increased risk of kidney and heart failure is also associated with early death in late-onset Fabry, and the soonest possible start to treatment can help slow or prevent such complications in both Fabry disease types.
Fabry disease in males and females
Fabry disease is an X-linked disease, meaning the mutated GLA gene resides on the X chromosome. This affects how the disease manifests in male versus female patients.
Men have only one X chromosome, inherited from their mother, that is active in all their cells. Men who inherit a mutated GLA gene from their mother will develop Fabry disease. Whether Fabry disease in males is characterized as classic or late-onset depends on the effects of the specific mutation and how much alpha-Gal A activity it allows.
While the specific GLA mutation also influences the disease type in women, the expression of Fabry disease in females is a bit more complex because they have two X chromosomes, each inherited from one of their biological parents.
In each cell of a woman’s body, one of those X chromosomes is randomly “turned off” and the genes it contains are not active, a natural process called X-inactivation. In the context of Fabry disease, this means that if a woman inherits a GLA mutation from one parent and a healthy GLA gene from the other, some of her cells will be healthy and produce a normal amount of functional alpha-Gal A, and others will have the mutated gene copy and won’t produce enough working enzyme.
Because the process is random, the proportion of healthy to mutated cells varies and can influence disease manifestation. For example, a higher proportion of healthy cells can partially compensate for the mutated ones and lead to milder disease.
This influence of X-inactivation means the presentation of Fabry disease is more variable in female patients. For example, some women with Fabry may have near normal alpha-Gal A levels and lack any obvious symptoms, even with a known Fabry-causing mutation in the GLA gene.
A girl with a GLA mutation associated with classic Fabry disease may see the same severity of symptoms as a boy with the same mutation or her symptoms might come on later and be milder. In this group of female patients, the typical starburst-like pattern of cloudiness in the eye’s cornea is commonly present.
Women with later-onset disease-causing mutations may never develop symptoms or may develop kidney or heart problems later in life. These women usually do not present with corneal findings or other symptoms associated with classic Fabry.
Fabry Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
-

-
 Discussion
Discussion
-

-

-
