
 Discussion
Discussion
Fabry disease overview
Last updated Aug. 19, 2024, by Lindsey Shapiro, PhD
 Fact-checked by Ana de Barros, PhD
Fact-checked by Ana de Barros, PhD
Fabry disease is a rare inherited condition characterized by the toxic accumulation of a fatty substance called globotriaosylceramide (Gb3) in the body’s cells. This buildup impairs organ function, especially in the kidneys and heart. The disease is also known as Anderson-Fabry disease, after the two physicians who independently discovered it in 1898.
Fabry occurs in men and women across racial and ethnic groups, affecting an estimated 1 in 1,000 to 9,000 people worldwide. Still, its prevalence is not well established, and it is believed that Fabry is underdiagnosed, especially in female patients.
No cure is currently available for the progressive disease, but there are treatments and interventions that can help slow its progression. If left untreated, or if a Fabry diagnosis is delayed, Fabry disease life expectancy is shorter than that of the general population — men with Fabry disease live, on average, into their late 50s, and women into their 70s.
Causes
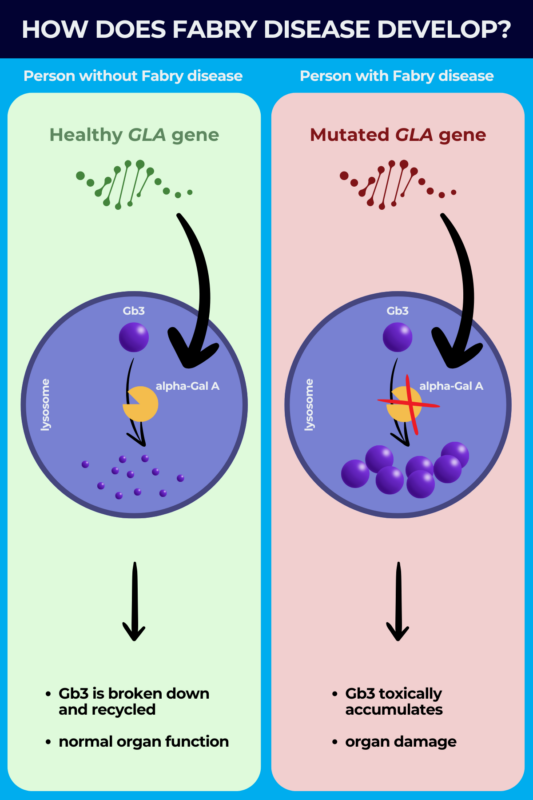
Fabry disease is caused by mutations in the GLA gene, located on the X chromosome, that contains the instructions for producing an enzyme called alpha-galactosidase A (alpha-Gal A).
Under normal conditions, alpha-Gal A is responsible for breaking down fatty molecules such as Gb3 inside a cellular compartment called the lysosome, where cell recycling processes take place.
The mutations, however, result in an alpha-galactosidase A deficiency that causes Gb3 to toxically accumulate in the lysosome. The condition is thus a lysosomal storage disorder, referring to a group of diseases where defects in one or more enzymes in the lysosomes cause substances to accumulate in cells.
Fabry disease inheritance happens in an X-linked dominant manner, meaning a person only has to inherit one mutated copy of GLA on a single X chromosome for Fabry to manifest.
Because women have two X chromosomes and men have only one, with the other being the Y chromosome, when a man with Fabry has children with a woman without it:
- all female children will inherit the mutated GLA gene
- no male children will inherit the disease, as they’ll only get a Y chromosome from their father.
When a woman with one mutated GLA gene has children with a man who doesn’t have Fabry disease, there is a 50% chance any of her children will inherit the mutated gene.
Affected men typically have a more standard disease course. For women, a healthy GLA gene on one X chromosome can partially compensate for a Fabry-causing mutation on the other due to a random cellular process called X-inactivation. Ultimately, this means that how Fabry manifests in women, and whether they have Fabry disease symptoms at all, is highly variable.
Types
There are two different types of Fabry disease — classic and late-onset. While both are caused by mutations in the GLA gene, they have a different disease course due to different amounts of functional alpha-Gal A enzyme being produced in each type. The amount of alpha-Gal A that’s made is informed by the specific disease-causing mutation.
Classic Fabry disease is the less common and more severe form of Fabry, characterized by virtually absent (less than 1% of normal) alpha-Gal A activity. Symptoms affecting the nervous system, skin, eyes, and gastrointestinal system usually emerge in childhood or adolescence and eventually progress to involvement of major organs such as the kidneys and heart.
Late-onset Fabry disease, the most prevalent form of Fabry, is characterized by greater than 1% of normal alpha-Gal A activity. Symptoms don’t usually start until adulthood, after the age of 30. The earliest symptoms seen in childhood during classic Fabry are often skipped with late-onset disease, and the first symptoms usually involve a major organ such as the heart or kidneys. While it overall progresses more slowly, late-onset Fabry can still lead to life-threatening organ damage in adulthood.
The presentation of classic or late-onset Fabry disease in female patients might look different than in males because, in addition to the type of mutation, the process of X-inactivation can influence how much alpha-Gal A is produced in the body.
Symptoms
The symptoms of Fabry disease arise from Gb3 accumulation in various tissues. Because it is a progressive disease, these symptoms tend to get worse over time. Still, the presentation of the disease can be highly variable from patient to patient and may not always follow a typical course, especially in female patients.
Common early Fabry disease symptoms, which are most often observed in people with the classic type of disease, include:
- nerve-related pain, usually manifesting as a burning pain in hands and feet
- angiokeratomas, a skin rash with clusters of small red spots on the skin
- corneal verticillata, a starburst-like pattern of cloudy areas on the eyes’ surface
- heat intolerance and decreased sweating
- gastrointestinal problems.
As the disease progresses, Fabry patients develop symptoms involving the body’s major organs, although this may be the first sign of disease for late-onset patients. Symptoms may include:
- heart disease, including problems with the heart’s pumping muscle, malfunctioning heart valves, abnormal heart rhythms, heart attack, or heart failure
- kidney dysfunction, which can eventually lead to kidney failure
- cerebrovascular disease, where blood flow in the brain or spinal cord is disrupted; stroke is a major cerebrovascular complication that can occur in Fabry.
Fabry disease kidney problems and heart complications are leading causes of premature death in Fabry patients.
While this is the standard disease course in male patients, Fabry disease symptoms in females may look very different. For example, a woman with a mutation associated with classic Fabry disease might not see the early childhood symptoms that would be expected in a male with the same mutation. Likewise, a woman with a late-onset Fabry-driving mutation may be largely without symptoms for most or all of her life.
Diagnosis
It is generally agreed that initiating treatment as early as possible gives patients the best chance of slowing disease progression and preventing or delaying life-threatening complications. As such, reaching a prompt and accurate Fabry disease diagnosis is essential for providing patients with the best possible outcomes.
Once Fabry disease is suspected based on clinical symptoms or family history, several diagnostic tests can help establish the diagnosis.
- Enzyme testing: This Fabry disease test measures the activity of the alpha-Gal A enzyme, where activity at 1% or lower is highly indicative of Fabry. This is most useful for diagnosing males with classic Fabry disease, as males with late-onset disease and female patients may have higher alpha-Gal A levels and will need additional testing.
- Genetic testing: This test looks for disease-causing mutations in the GLA gene, and is the most reliable way to diagnose Fabry in all patients. Fabry disease genetic testing can also be used to identify the disease in other family members once a person is diagnosed.
- Lyso-Gb3 test: Lyso-Gb3, a metabolite of Gb3, is measured in a body fluid or tissue sample, and may be elevated in Fabry. This test may be useful for supporting a Fabry disease diagnosis when genetic testing results are not clear or in female patients without symptoms.
- Prenatal testing and newborn screening: These tests can be used to look for GLA mutations or measure alpha-Gal A enzyme activity in an unborn fetus or a newborn baby to determine if they will be affected by the condition.
- Other tests: A range of tests, including cardiac evaluations, brain MRI scans, and blood or urine tests to determine kidney function might be used when doctors are establishing Fabry and ruling out differential diagnoses. Such tests may also be used to monitor disease progression.
Treatment
While there is yet no cure for Fabry disease, treatments are available to slow disease progression and help manage symptoms to make life more comfortable for people with Fabry. There are two main types of Fabry disease treatments:
- Enzyme replacement therapy (ERT) works to deliver a functional, lab-made version of the alpha-Gal A enzyme directly into the bloodstream. This allows the body to more effectively break down Gb3, which eases disease symptoms and delays the appearance of life-threatening disease complications. Two ERTs are approved in the U.S. for Fabry disease regardless of disease-causing mutation: Fabrazyme (agalsidase beta) and Elfabrio (pegunigalsidase alfa-iwxj).
- Chaperone therapy, a newer class of oral Fabry disease treatment, works to restore the function of abnormal alpha-Gal A versions that are produced from certain GLA mutations. Galafold (migalastat) is the only chaperone therapy approved in the U.S., and is indicated for patients who carry mutations expected to be responsive to chaperone therapy.
Fabry patients might also be treated with a wide range of other therapies to help manage specific symptoms. This may include:
- medications to control blood pressure, heart rhythm, or to prevent blood clots for managing heart disease
- oral medications, dialysis, or kidney transplant for kidney issues
- blood thinners to lower the risk of stroke
- medications or lifestyle adjustments to ease and manage pain
- treatments to ease diarrhea, nerve-related abdominal pain, gastrointestinal inflammation, nausea, or other gastrointestinal problems
- laser treatment for angiokeratomas
- hearing aids or surgery to manage hearing loss.
Because Fabry disease is caused by genetic mutations, there are no known treatments or lifestyle changes that are effective for Fabry disease prevention. Fabry treatments can, however, help to slow disease progression and possibly delay or prevent the onset of disease complications for as long as possible.
Living with Fabry disease
Navigating life with Fabry disease can be stressful and may come with many challenges. Staying informed about the disease and proactively managing physical and emotional changes as they arise can help patients have the best possible quality of life.
A few things a person living with Fabry can do to manage the disease include:
- Establishing a multidisciplinary care team: A well-rounded care team will help monitor disease progression and decide the most effective treatment plan.
- Avoiding triggers: Overactivity and fatigue, excess heat, stress, and illness or injury can be triggers for Fabry-associated pain. Avoiding them may help prevent pain and ensure patients retain their ability to function.
- Exercising and working with a physical therapist: While excess exercise can be a trigger for pain in Fabry, a regular, low-impact exercise program approved by one’s care team can help patients maintain physical and mental health.
- Making recommended diet and nutrition changes: There’s no specific Fabry disease diet, but certain dietary changes, made in consultation with the patient’s care team, can help ease gastrointestinal issues. This could include having smaller and more frequent meals, staying hydrated, and limiting alcohol or foods that could upset the stomach like fried, greasy, or spicy items.
- Seeking mental health support: All of the above factors can make life with Fabry easier, supporting a patient’s mental health. In some cases, a counselor or psychologist might also help patients work through emotional struggles and come up with coping strategies to deal with stress.
Establishing a network of family, friends, healthcare professionals, and other members of the Fabry community are also an important aspect of disease management. Patients can find online or in-person Fabry disease support groups to help them build their support network.
Other online resources that can offer information, tips, and support include:
Fabry Disease News also provides patients and caregivers with a dependable source of up-to-date information and news on Fabry disease, as well as insightful columns that share personal stories from people living with the condition.
Fabry Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
Discussion